Writer: Naomi Choi
Editor: Lorrine Sum
Artist: Yasmin Yong
Most people have heard of Huntington’s disease, but some might not realise how severely it could affect the brain. In fact, Huntington’s disease is one of the most difficult-to-treat inherited brain disorders and is characterised by its progressive symptoms, gradually impacting everything from memory to behaviour. Current treatments for Huntington’s focus on managing and minimising symptoms, rather than targeting its root cause. For decades, patients and families have hoped for a therapy that could do more than simply manage the condition, and a research team at UCL has taken the first major step towards changing the future of Huntington’s treatment. To understand why this research breakthrough matters, let’s take a step back to look into the disease itself and what makes it so challenging to treat in the first place.
Understanding Huntington’s Disease
Huntington’s disease (HD) is a fatal, inherited neurodegenerative disorder caused by a single mutation in the huntingtin (HTT) gene, leading to the progressive loss of neurones and symptoms including cognitive decline, motor impairment and behavioural changes. The HTT gene in Huntington’s disease is mutated to contain an expanded CAG trinucleotide repeat, leading to the production of faulty huntingtin mutants. While the normal huntingtin protein is harmless and in fact plays important roles in cell survival, gene regulation and early development of the brain, its mutant version is toxic since its misfolding and accumulation result in the formation of insoluble protein aggregates known as inclusion bodies within neurones. Over time, these harmful clumps of huntingtin mutants disrupt essential cellular processes, such as transcription, mitochondrial function, autophagy and synaptic signalling, ultimately triggering neuronal dysfunction and cell death. It is important to know that neurones do not replicate; thus, once a neuronal cell dies, it will not be replaced.
The region in the brain that is most affected is the basal ganglia, which plays key roles in controlling movement, cognition and behaviour. As neurones in the basal ganglia degenerate progressively, affected individuals begin to develop the characteristic symptoms of HD, including involuntary movements, impaired memory and coordination, as well as mood disturbances and personality changes. These symptoms could eventually lead to physical and psychological complications such as respiratory infections, heart failure, impaired swallowing (dysphagia) and movement (chorea), fatigue, depression, anxiety, agitation and more, with the most common cause of death being aspiration pneumonia that is often caused by dysphagia.
Huntington’s disease follows an autosomal dominant inheritance pattern, meaning that people with an affected parent have a 50% probability of inheriting the disease, with first symptoms typically arising between the ages of 30-50, and worsening over 10-25 years after symptoms first develop. Eventually, affected individuals are confined to bed and require total care, and the disease is normally fatal within two decades. Around 75,000 people across the UK, US and Europe are currently living with HD, with many more at risk of developing it.
UCL-Led Gene Therapy for Huntington’s Disease
Early diagnosis and treatment is key to treating HD, yet no treatment has been able to slow, prevent, or halt disease progression – not until now. A pioneering clinical trial led by researchers from the UCL Huntington’s Disease Research Centre has revealed that a new gene therapy has shown promising results in slowing disease progression in HD patients.
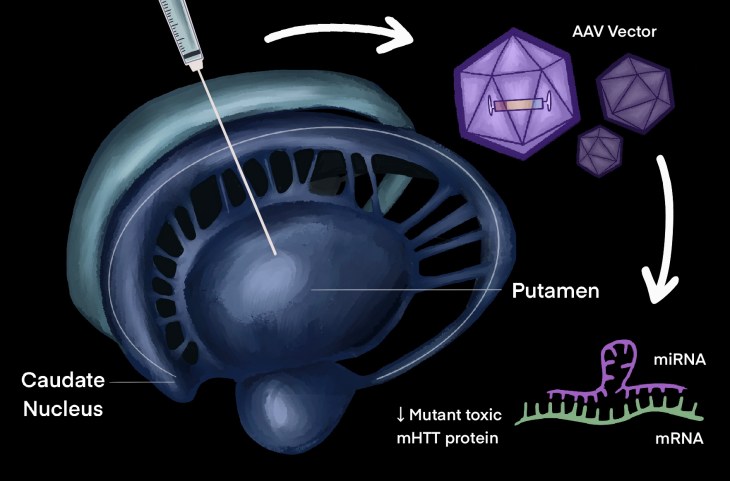
Prof. Sarah Tabrizi, director of the research centre and lead scientific advisor on the clinical trial, and Prof. Edward Wild, principal investigator of the UCL Huntington’s Disease Centre trial site, led the development of the gene therapy named AMT-130. The gene therapy is delivered through a one-off neurosurgery by injecting an adeno-associated viral (AVV) vector into the brain using real-time MRI scanning to guide a micro-catheter to the caudate nucleus and the putamen, which are the two regions of the basal ganglia most vulnerable to HD. The viral vector contains a DNA sequence that codes for a small fragment of microRNA (miRNA), which performs gene silencing by binding to the section of messenger RNA (mRNA) encoding the huntingtin mutant (mHTT) protein in neurones. This marks it for degradation, thus switching off its production and reducing mutant protein level.
AMT-130’s clinical trial was developed by uniQure, a gene therapy company based in the Netherlands, and involved 29 patients who had tested positive for the HD gene and were diagnosed with early-stage HD. These patient volunteers have completed up to 36 months of Phase I/II clinical trials, with 12 receiving high-dose therapy. Results showed a 75% slowing of clinical progression compared to a matched external cohort of HD patients, as measured by the composite Unified Huntington’s Disease Rating Scale, as well as a decreased level of neurofilament light protein (NfL), a biomarker protein released into the spinal fluid when neurones are damaged. The treatment was also considered generally safe and well tolerated, and a single administration should last for life as neurones do not regenerate and get replaced.
The Future of Huntington’s Treatment
This breakthrough with the AMT-130 gene therapy undoubtedly marks a significant and exciting shift in how researchers approach Huntington’s disease, offering the first real possibility of slowing down the condition at its genetic roots while opening the doors to treating other neurodegenerative conditions with gene therapy – a milestone that began right here at UCL. Although full clinical trial results are yet to be published and larger-scale studies might be required to confirm long-term safety and effectiveness, uniQure is already working toward securing FDA approval, reflecting strong confidence in APT-130’s therapeutic potential. As research progresses, the ultimate hope is that Huntington’s disease, once considered untreatable and inevitably fatal, could one day be met with therapies that fundamentally change its course and profoundly improve the lives of hundreds of thousands of people affected worldwide.
